Projects
-

Cheminformatics & Artificial Intelligence
Focus: We develop new computational algorithms for ligand- and structure-based drug design. We are actively engaged in creating new chemical embedding strategies and neural network architectures for quantitative structure-activity/property relationship (QSAR/QSPR) modeling, protein-ligand binding affinity estimation, and high-throughput induced-fit protein-ligand pose refinement.
Impact: Traditional experimental approaches to drug discovery, while accurate, are time-consuming and expensive. Because of its ability to complement experimental methods and reduce costs, computer-aided drug design (CADD) has become an essential component of the drug discovery process. Unfortunately, however, computational methods remain limited in their accuracy, generalizability, and throughput.
Image generated from PDB ID 6DAR. -

RosettaQM
Focus: In collaboration with our friends at the Flatiron Institute, we develop RosettaQM, an extension of the Rosetta macromolecular modeling and design software suite that enables quantum mechanics (QM) calculations.
Impact: Accurate modeling of biomolecular systems is essential for understanding biological processes at the molecular level. Quantum Mechanics (QM) offers detailed insights into the electronic structures of molecules, which are paramount for predicting reactivity and interaction energies accurately. RosettaQM bridges the gap between the computational efficiency of molecular mechanics and statistical energy functions and the accuracy of quantum chemistry. It integrates QM calculations within the Rosetta biomolecular modeling suite, enabling users to perform hybrid simulations. By incorporating QM calculations into biomolecular modeling, RosettaQM enhances our ability to design covalent inhibitors, model enzymes, and understand disease mechanisms at an atomic level.
-

Partial Agonist Design for the Mu Opioid Receptor
Focus: We utilize an extensive repertoire of computational tools to identify µ opioid receptor (µOR) partial agonists as potential pharmacological agents to help treat addiction and manage pain with reduced side effects. We are also broadly interested in understanding the determinants of µOR ligand intrinsic activity, allostery, and selective G-protein recruitment, as well as the potential role off-target opioid receptor activation in treating addiction and managing pain. To accomplish our goals, combine ligand-based QSAR modeling and pharmacophore modeling, structure-based docking, and long-timescale molecular dynamics (MD) simulations. We experimentally evaluate our predictions through collaboration with academic and commercial groups.
Impact: The ongoing opioid crisis demands new treatment strategies to help patients with recovery, and patients deserve pain relief options that do not lead to addiction. Partial agonists of the µOR present a promising avenue. Thus, developing next-generation µOR partial agonists could significantly alter pain management strategies to address a critical public health issue.
-
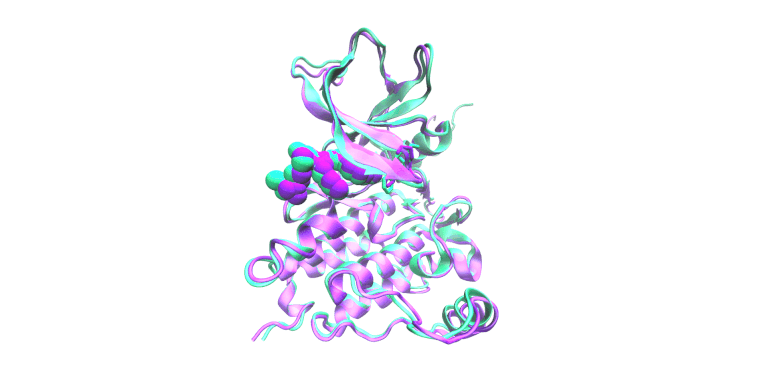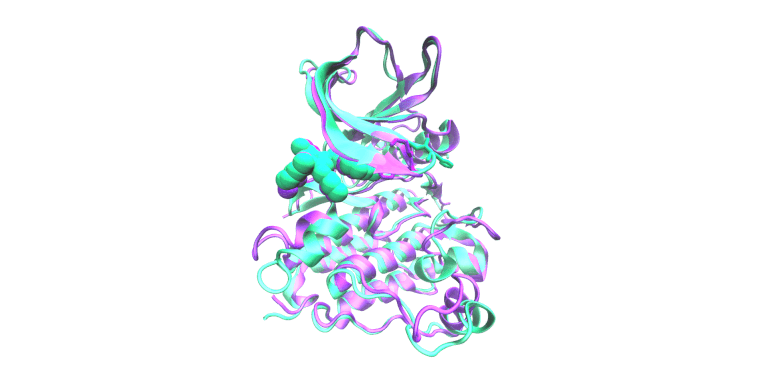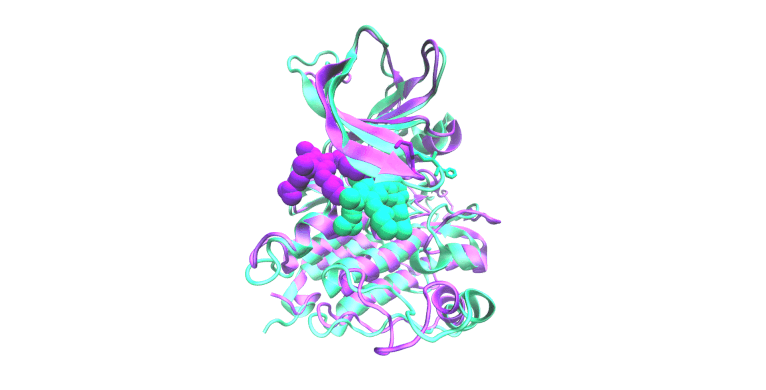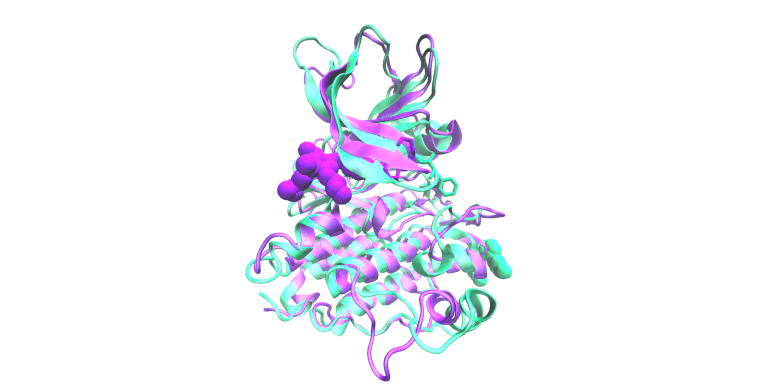
Molecular Simulation & Precision Oncology
Focus: We employ all-atom molecular dynamics simulations to investigate the structural and functional impacts of oncogenic mutations on epidermal growth factor receptor (EGFR) kinase family proteins. These simulations provide detailed insights into how mutations alter receptor conformation and activity, guiding clinical decision-making and informing the design of new therapeutic strategies. We collaborate extensively with the laboratories of Christine Lovly, MD, PhD and Adam Smith, PhD for these studies.
Impact: Oncogenic mutations in proteins like EGFR are central to cancer development and progression. Understanding these mutations at an atomic level is crucial for developing targeted therapies.
The display simulation shows osimertinib dissociating from EGFR E746_S752>V/G724S (green) but not E746_S752>V (pink).